研究者の方へ

千葉大学大学院医学研究院
アレルギー・臨床免疫学
千葉大学病院
アレルギー・膠原病内科
アレルギー・臨床免疫学では、アレルギー疾患や自己免疫疾患の基礎研究と臨床研究を行っています。「すぐに」とまでは言いませんが、遠からず臨床に役立つ可能性のある研究に重点を置いています。個々のメンバーの興味をできるだけ尊重しつつ、それぞれの研究が有機的に結びつくように心掛けています。
将来的には、1)アレルギー疾患の本質である非侵襲的抗原(アレルゲン)に対して過剰な免疫応答が惹起される分子機構の解明とその制御法の開発、2)自己抗原に対する免疫応答機構の解明とそれに基づく自己免疫疾患の治療法の開発を行ないたいと考えています。研究という禁断の果実を一緒に味わいませんか?
喘息患者の約10%を占める難治性喘息にはこれらのTh2細胞、Th2サイトカインを標的とした治療方法の効果が乏しく、異なる病態が関与していることが示唆されていました。我々の研究室では難治性喘息患者の気道で好中球性炎症が惹起されていること、さらには気道でのIL-17産生が喘息の重症度と相関することに着目し、Th17細胞がTh2細胞依存的に惹起されるアレルギー性炎症を増悪させることを示しました(Wakashin, Am J Respir Crit Care Care Med 2008)。また、このTh17細胞分化の分子機構に関して、肺のCD11b陽性樹状細胞に発現するDectin-2が必須であることを見出しています(Norimoto, Am J Respir Cell Mol Biol 2014)。このようにDectin-2/Th17経路が喘息の増悪に関わることを示す一方、喘息の抑制系にも着目しIL-22が気道上皮細胞からのIL-25産生を抑制することによりアレルギー性気道炎症を制御していることを示しています(Takahashi, J Allergy Clin Immunol 2011)
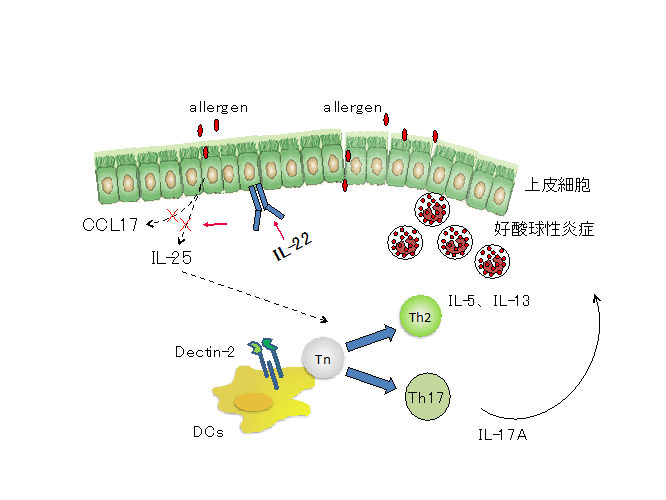
気道上皮細胞は、物理的バリアとして機能するだけでなく、アレルゲン、ウイルス、大気汚染物質等の外界からの様々な刺激に応答して種々のケモカインやサイトカインを分泌することで、樹状細胞や2型ヘルパーT(Th2)細胞だけでなく2型自然リンパ球(ILC2)を活性化し、喘息の病態において重要な役割を果たしていることが明らかにされています。気管支喘息の治療では、従来の吸入ステロイドに加え、IgEやTh2サイトカインをターゲットにした生物学的製剤が使えるようになりましたが、依然として数%の患者は治療抵抗性の難治性喘息です。
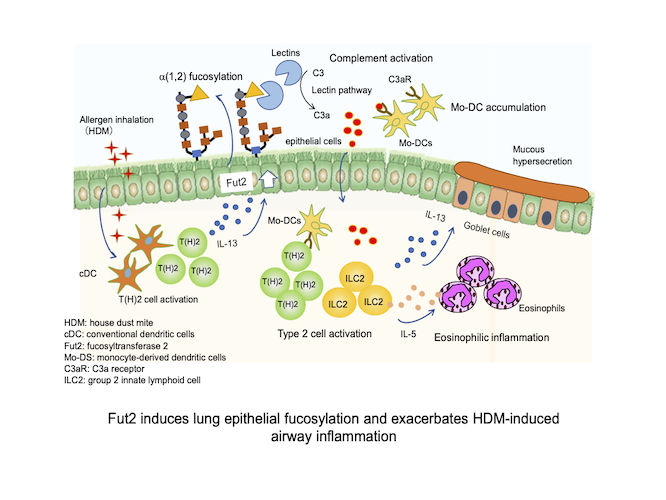
私たちは難治性喘息の克服を目指し、アレルギー性気道炎症における気道上皮細胞の役割に着目して研究を行なっています。最近の成果として、IL-22が気道上皮におけるReg3γ(抗菌ペプチドの一種)産生を誘導し、サイトカイン産生の抑制を介してアレルギー性炎症の進展を抑制すること(Ito T et al., J Exp Med 2017)、フコース転移酵素のFut2が気道上皮のフコシル化を誘導し、補体の活性化と樹状細胞(DC)の集積により気道炎症が悪化すること(Saku A et al., J Allergy Clin Immunol 2019、Allergy 2020)を明らかにしました(図)。
引き続き、気道上皮細胞と免疫細胞とのクロストークに着目し、新たな治療ターゲットを開拓することを目標としています。
関節リウマチは原因不明の慢性関節炎です。その病態解析は炎症が生じるメカニズムに関する研究を中心に活発に行われてきました。その成果として炎症性サイトカイン等をターゲットとした生物学的製剤やJAK阻害薬(b/tsDMARDs)が開発され、文字通りゲームチェンジャーとなりました。我々もこれまでリウマチの悪化に関わる因子について解析を行ってきました(Shoda et al., Arthritis Res Ther. 2022, Etori et al., Rheumatology. 2023, Suga et al., JCI insight. 2023)。しかし、b/tsDMARDsを使用中にもかかわらず再燃を繰り返す症例を経験するため、新たな着眼点に基づく治療戦略が必要と言えます。
現在、関節炎症の収束および再燃というフェーズに着目し、そこでの細胞分子機構を解明することによる新規治療の基盤開発を志しています。
その他、組織に在住する制御性T細胞についての研究にも興味を持っています(Tanaka et al., J Exp Med. 2019, Kasuya et al., Sci Rep. 2023)。

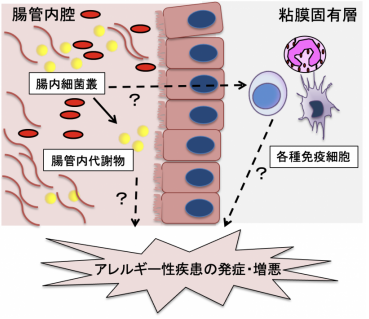
ヒトを含め動物の腸内には100兆個に及ぶ細菌が生息しており、これらの一群を腸内細菌叢と呼ばれています。腸内細菌叢や腸内細菌叢が産生する腸管内代謝物は以前より炎症性腸疾患、関節リウマチなどの自己免疫疾患、糖尿病・高血圧などの生活習慣病との関連が指摘されております。アレルギー性疾患においても代表的な腸管内代謝物の一種であるプロピオン酸およびGPR41陽性好酸球がアレルギー性気道炎症を抑制するメカニズムが報告されております(Ito et al., Gut microbe. 2023)。
私達は腸内環境(腸内細菌叢・腸管内代謝物)がアレルギー性疾患の発症・増悪に寄与するメカニズムの解明を目指しています。
免疫疾患は遺伝的な要因と後天的な要因の相互作用により発症すると考えられていますが、早期発症、重症、家族歴がある患者さんなどの中に一定数、遺伝的な要因が強い患者さんがいると考えられております。近年、家族性地中海熱などの自己炎症性疾患においては保険診療で遺伝子検査が可能になりましたが、免疫疾患における遺伝子診療は発展途上です。私たち遺伝学チームでは、2022年より遺伝性免疫異常症が疑われる患者さんとそのご家族に対して全エクソーム配列解析による原因遺伝子の同定を臨床研究として開始しました(図1)。その結果、単一遺伝子変異により発症していると考えられるいくつかの疾患を同定致しました。単一遺伝子の変異によって引き起こされる先天性免疫異常症患者を同定・研究することは、診断を超えてヒトにおける免疫制御の分子メカニズムや免疫疾患の発症メカニズムに新しい発見をもたらします。私たち遺伝学チームは患者の変異を持つ遺伝子組み換えマウスの作成や分子生物学的解析、免疫学的解析などの基礎的研究も行い、免疫疾患の分子メカニムの解明と治療法の開発を行っております。
<診断実績>
家族性地中海熱を含む自己炎症性疾患、慢性肉芽腫症、家族性血球貪食症候群、外胚葉形成不全症、GATA2異常症、補体欠損症、単一遺伝子変異による全身性エリテマトーデス

我々はIL-17Aの主要なシグナル伝達分子であるNF-kB1がImiquimod誘導性乾癬モデルにおいて重要な役割を果たしていることを見出した。さらにNF-kB1欠損マウス由来のケラチノサイトをIL-17Aで刺激し、刺激前後の遺伝子発現変化を網羅的に解析したところ、分子Xが強く発現誘導されることを見出した。現在、乾癬患者皮膚組織における分子Xの発現レベルの解析と分子Xの遺伝子欠損マウスの樹立と同マウスにおける乾癬モデルを解析中である。
